 |
 |
 |
Inferring protein structure and dynamics from simulation and experiment
Kyle A. Beauchamp
Das and Pande Labs
Biological Macromolecules
The Atomistic Basis of Disease
...EVKMDAEFRHDS... ...EVKMDTEFRHDS... ...EVKMDVEFRHDS...
A crisis at the atomic scale
Goal: Predictive, atomic-detail models of protein structure and dynamics
Introduction to Molecular Dynamics
- Simulate the physical interactions of proteins in solution
- Numerically integrate the equations of motion
100 $\mu s$ of HP35 Dynamics
Goals of Atomistic Simulation
- Calculate experimental observables
- Generate atomic-detail hypotheses
- Interpret experimental observables
Outline
- Inferring Protein Dynamics from Molecular Simulation
- Quantitative Comparison of Villin Headpiece Simulations and Triple-Triplet Energy Transfer Experiments
- Inferring Conformational Ensembles from Noisy Experiments
Inferring Protein Dynamics from Molecular Simulation
Two Challenges in Molecular Simulation
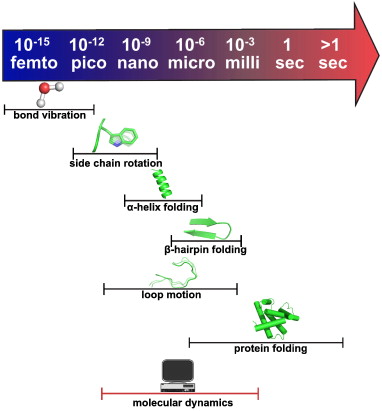
How to sample biological timescales?
Two Challenges in Molecular Simulation
How to sample biological timescales?


Two Challenges in Molecular Simulation
Meaningful Connection to experiment
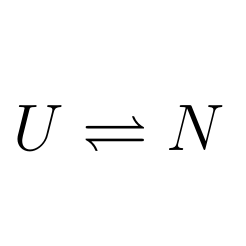
Markov State Models
- Run many parallel simulations on commodity hardware
- Build kinetic network model of dynamics
- Quantitatively predict structure, equilibrium, and kinetics
100 $\mu s$ of HP35 Dynamics (MSM)
Introduction to Markov State Models
Introduction to Markov State Models
Introduction to Markov State Models
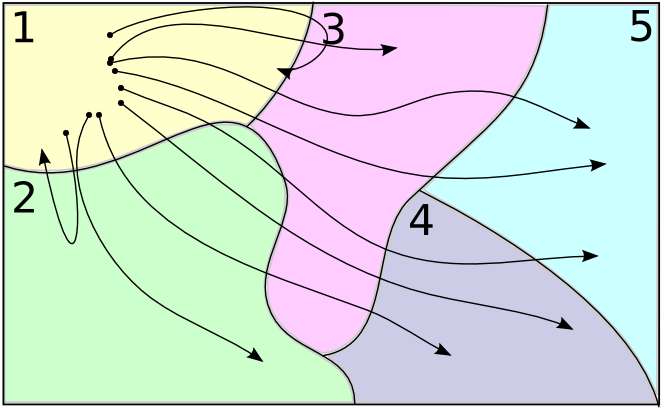
Counting Transitions
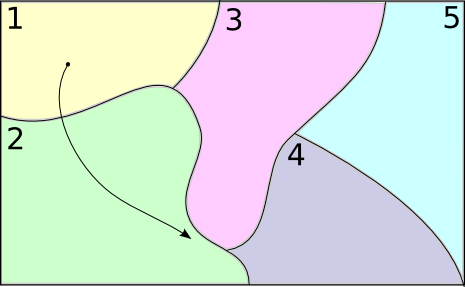
$\downarrow$
$A = (111222222)$
Counting Transitions
$A = (111222222)$
$\downarrow$
$C = \begin{pmatrix} C_{1\rightarrow 1} & C_{1\rightarrow 2} \\ C_{2 \rightarrow 1} & C_{2 \rightarrow 2} \end{pmatrix} = \begin{pmatrix}2 & 1 \\ 0 & 5\end{pmatrix}$
Estimating Rates
$\downarrow$
$T = \begin{pmatrix} T_{1\rightarrow 1} & T_{1\rightarrow 2} \\ T_{2 \rightarrow 1} & T_{2 \rightarrow 2} \end{pmatrix} = \begin{pmatrix}\frac{2}{3} & \frac{1}{3} \\ 0 & 1\end{pmatrix}$
MSMBuilder
An Open-Source, High-Performance, Python toolkit for MSM Construction
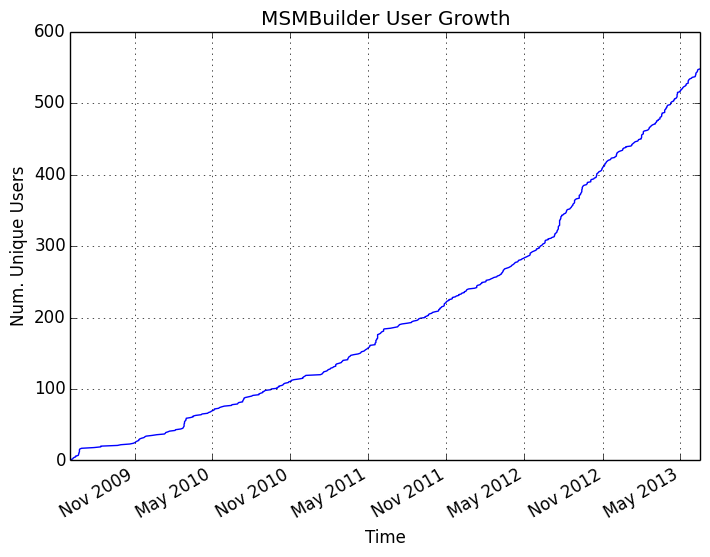
Progress in MSM Construction
Scaling to full protein systems
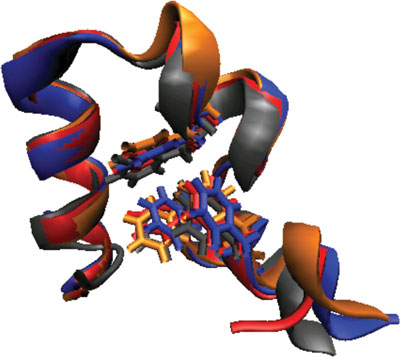
Progress in MSM Construction
Reaching millisecond timescales
Progress in MSM Construction
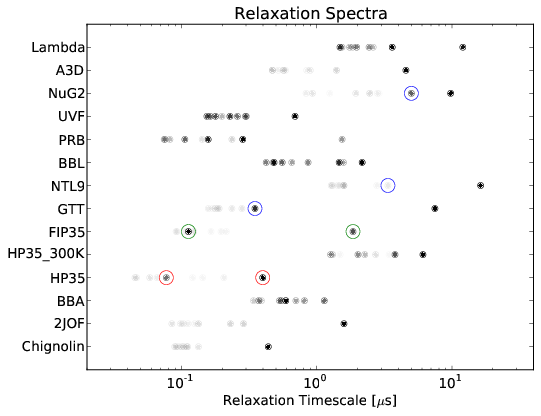
Algorithms for Accurate MSMs
Progress in MSM Construction
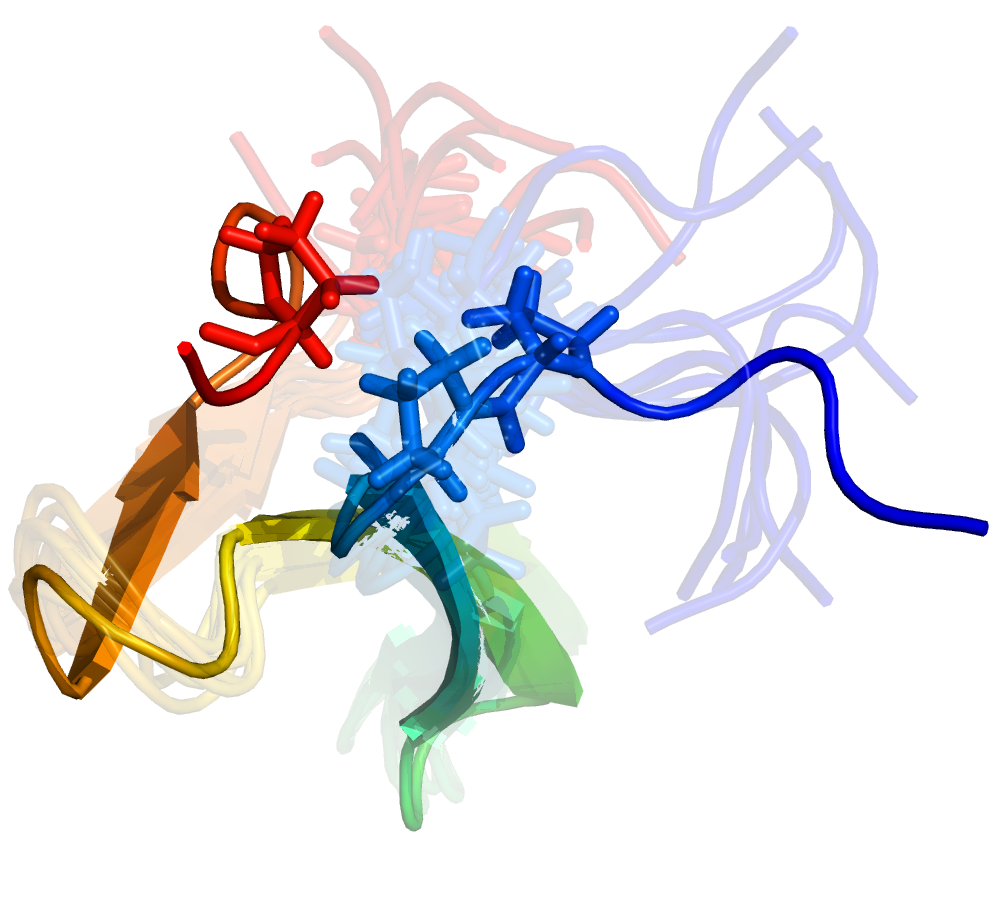
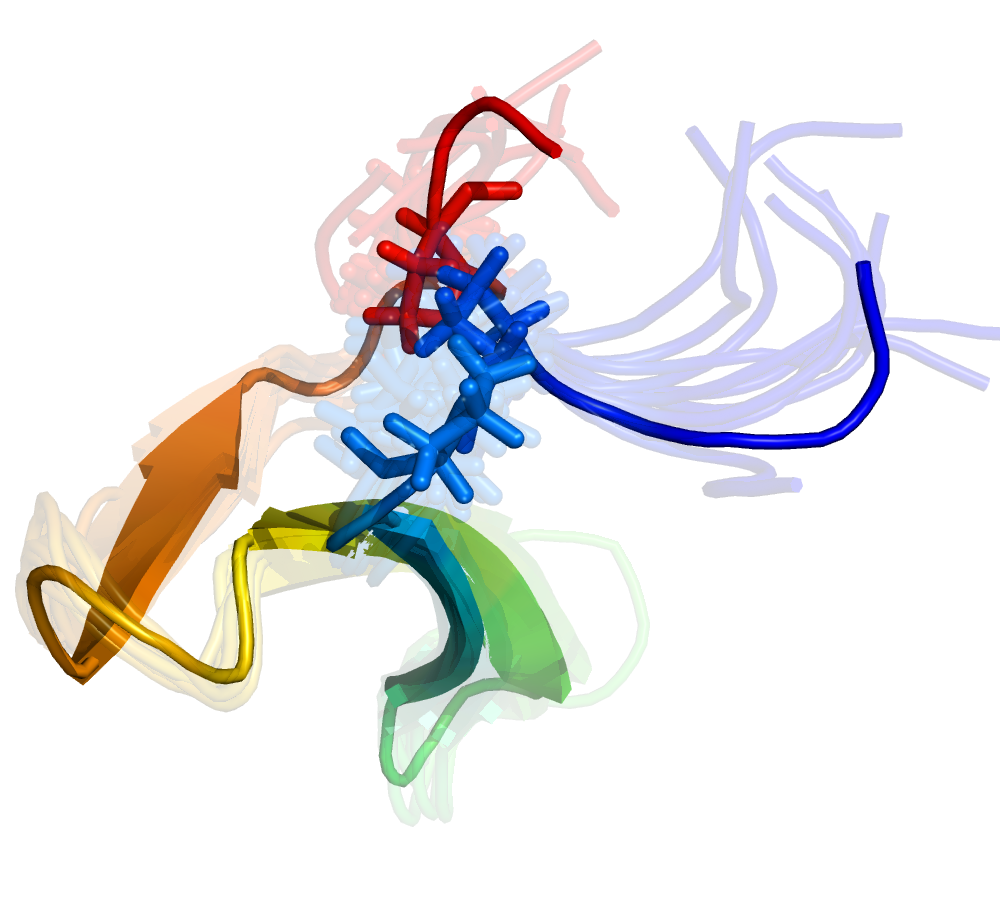
Reconciling two and multi-state behavior in protein folding
Quantitative Comparison of Villin Headpiece Simulations and Triple-Triplet Energy Transfer Experiments
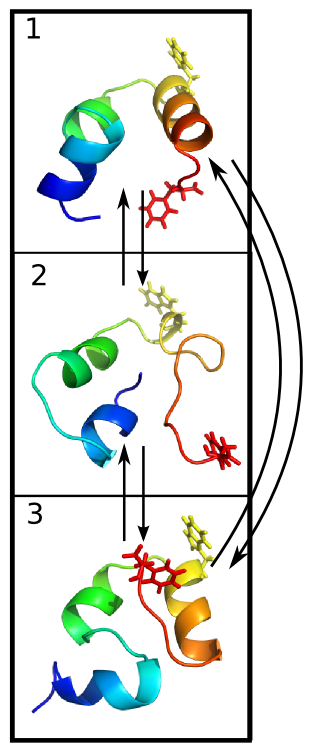
HP35: A Model for Protein Folding
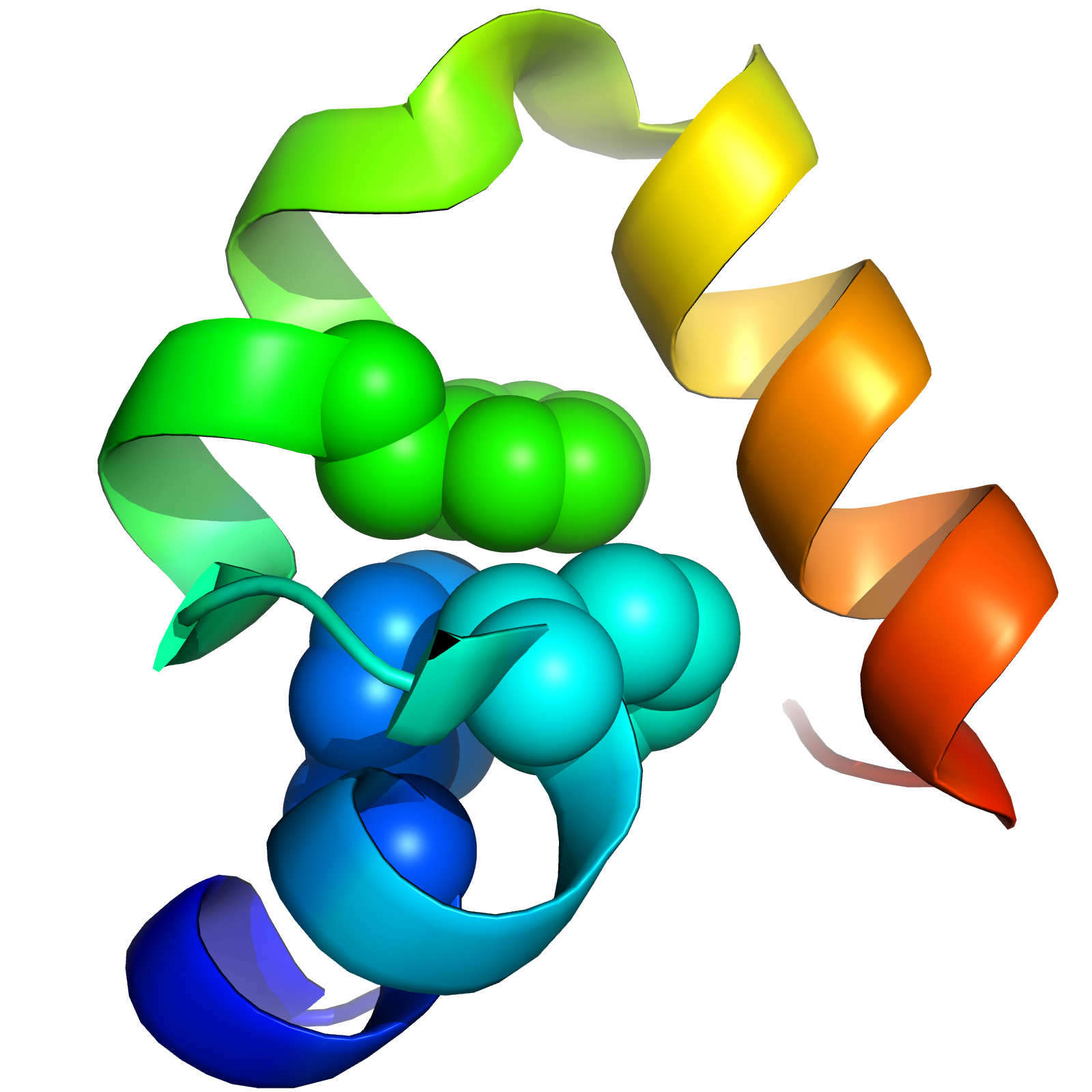
Triplet Triplet Energy Transfer
- Like FRET, but sensitive at the Å scale
- Used to monitor rates of contact formation
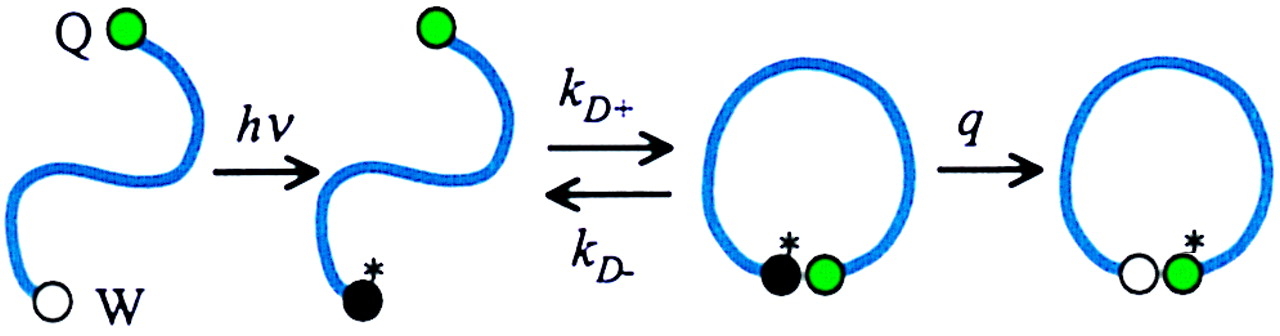
Triplet Triplet Energy Transfer
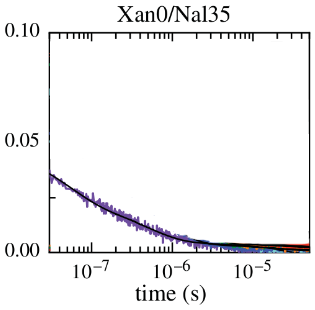
Observed TTET at 4 probe locations
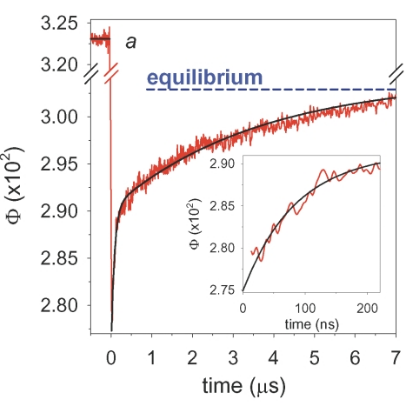
Simulating TTET in Silico
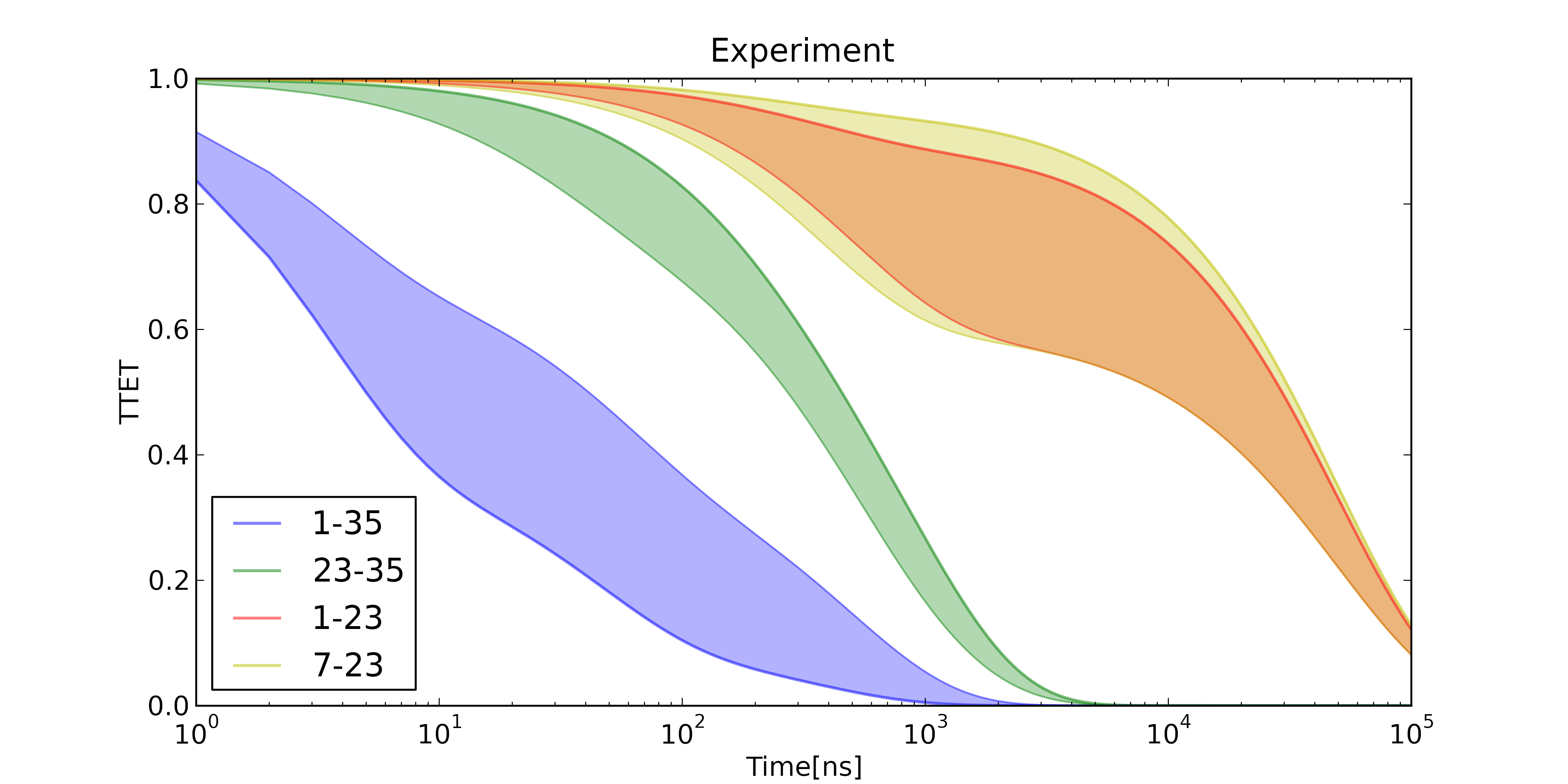
A modified MSM can predict the output of TTET experiments
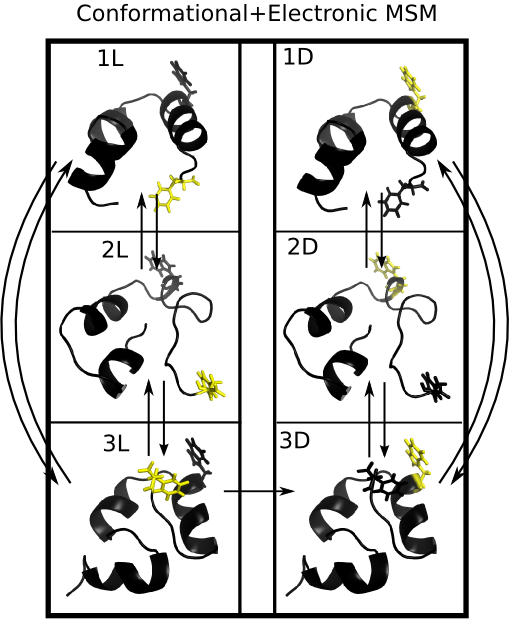
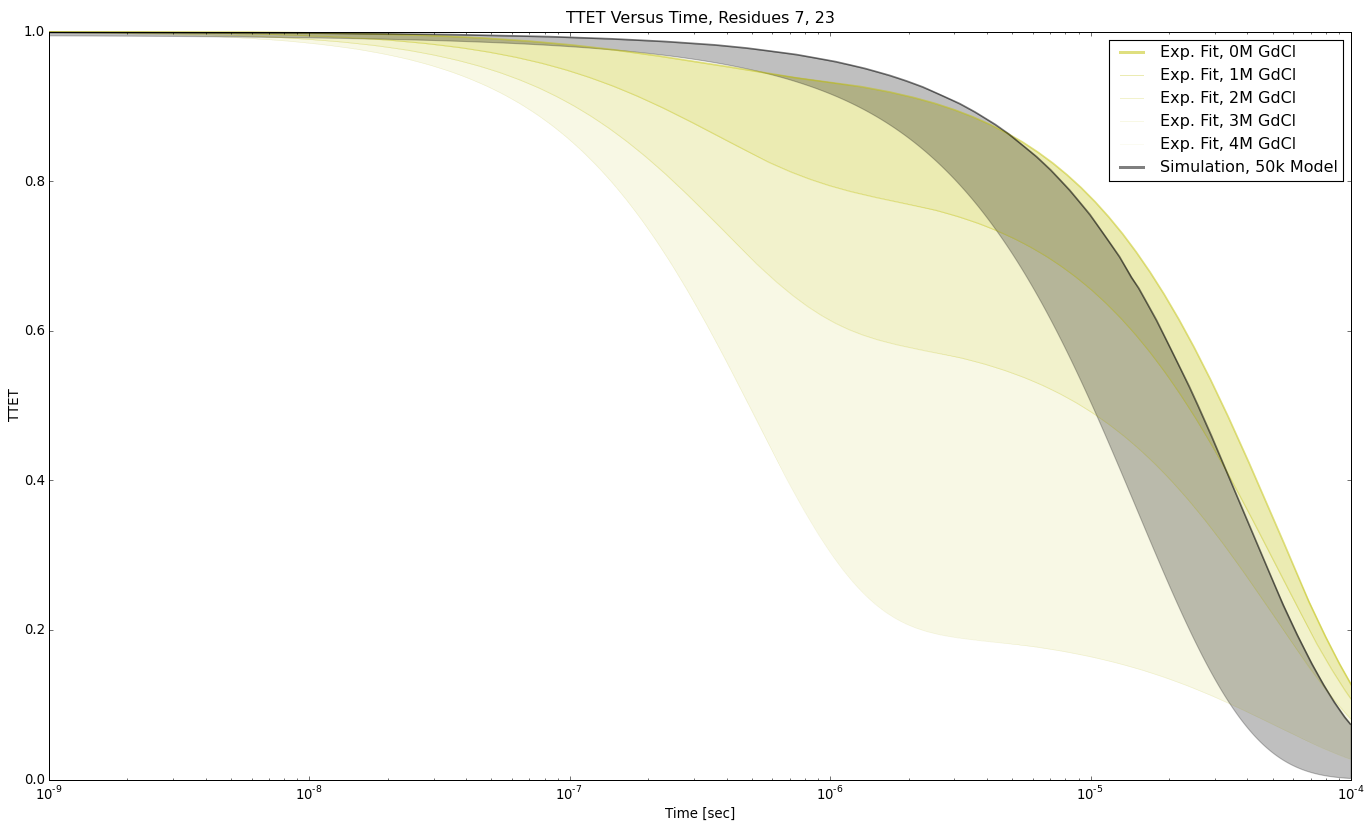
TTET at positions (7,23)
TTET at positions (7, 23)
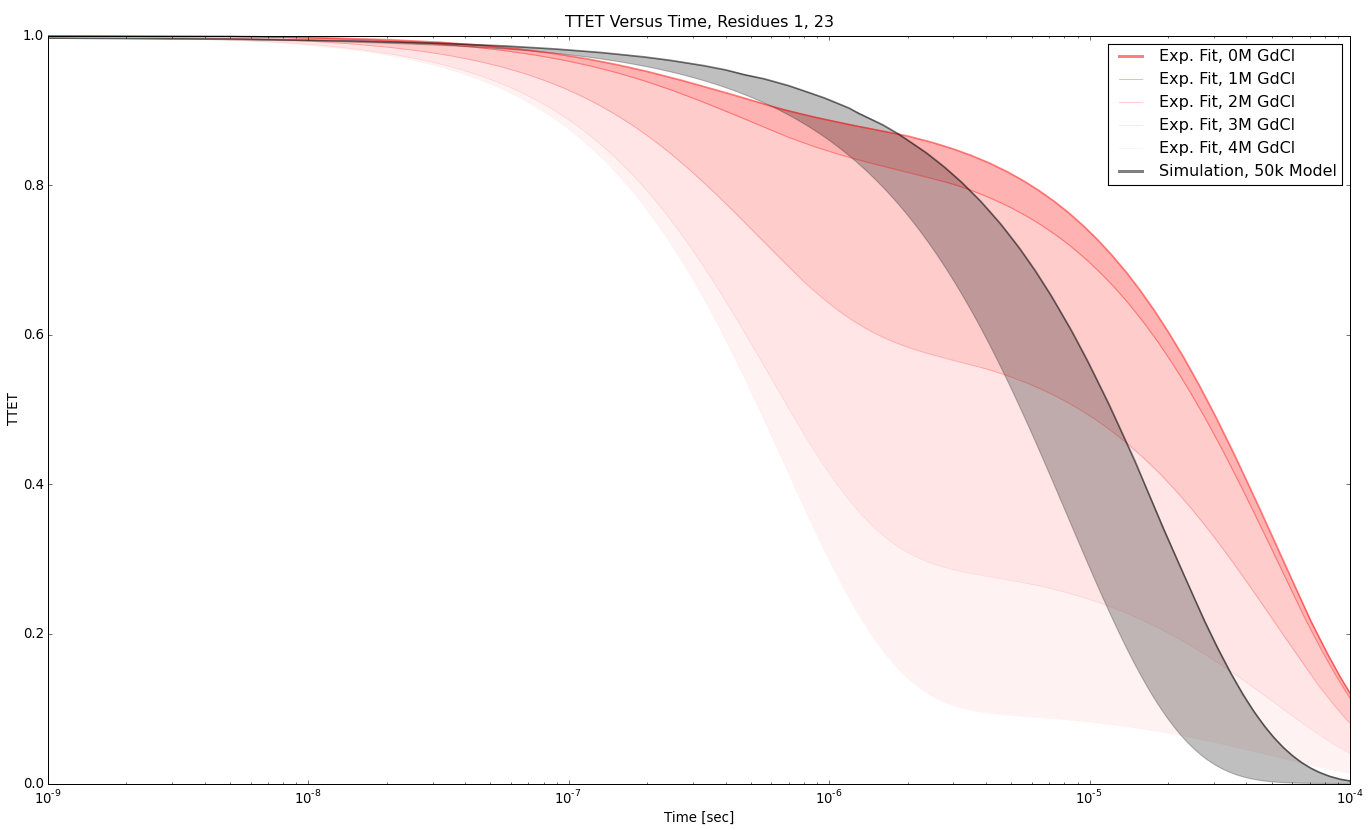
TTET at positions (1, 23)
TTET at positions (1, 23)
TTET at positions (23, 35)
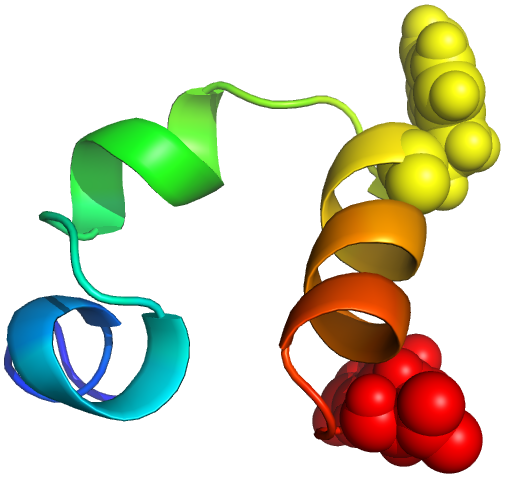
TTET at positions (23, 35)
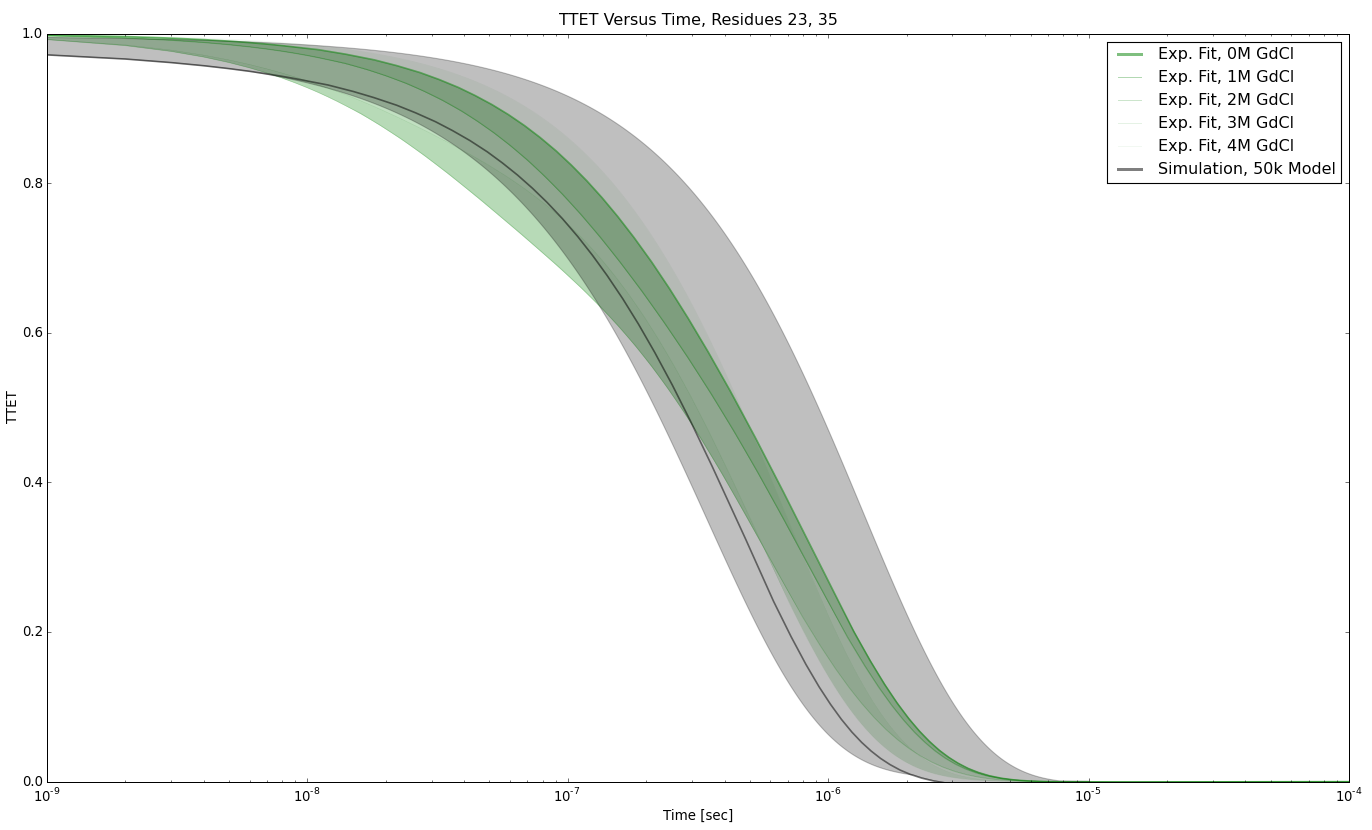
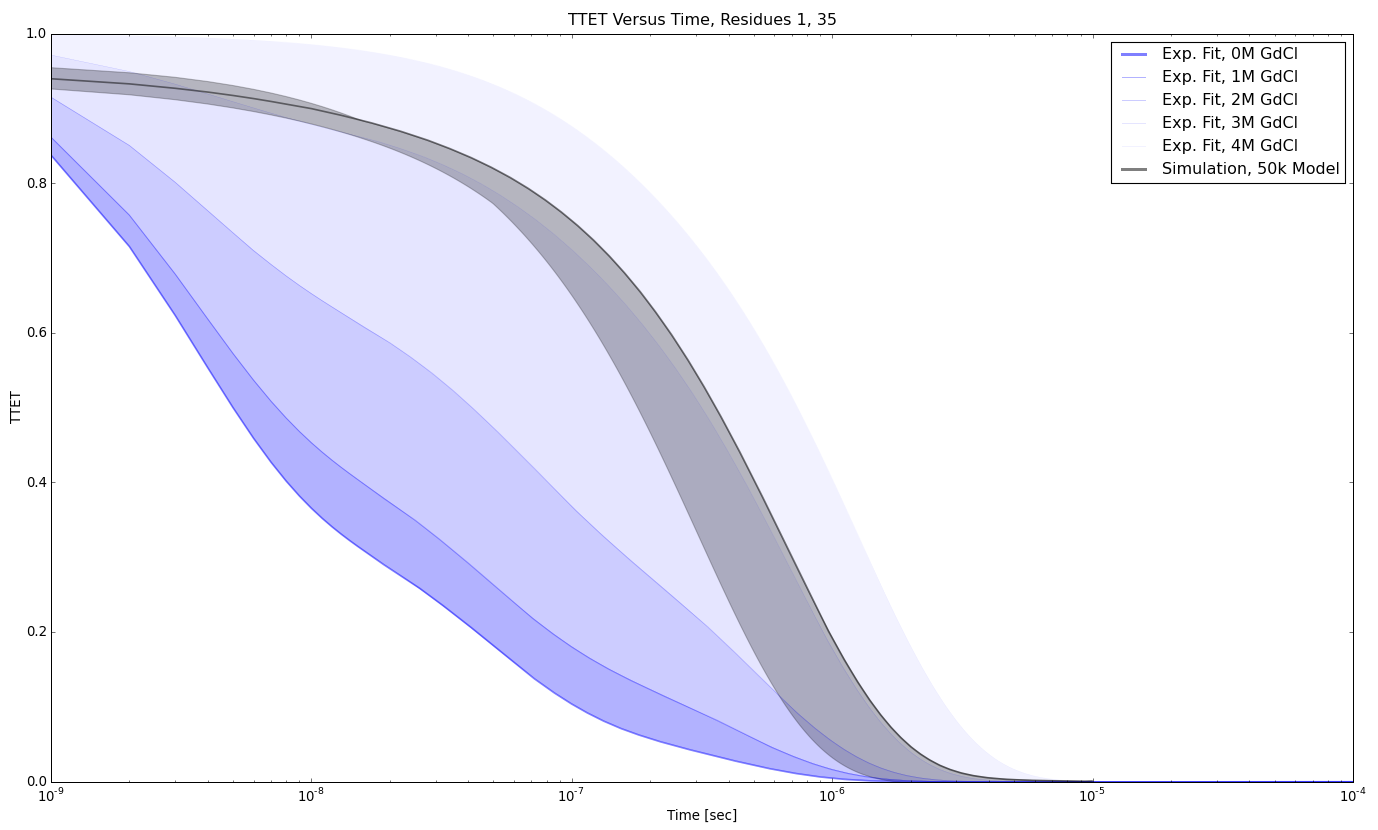
TTET at positions (1, 35)
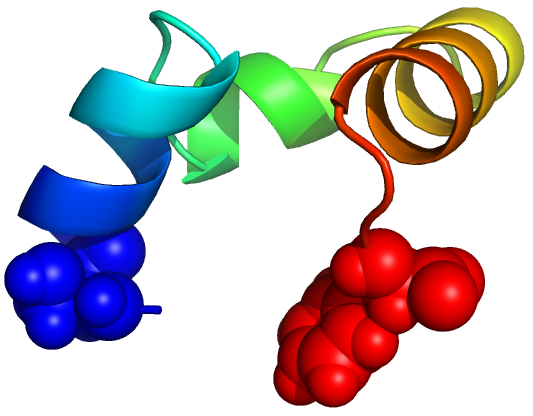
TTET at positions (1, 35)
Predicting All TTET Experiments
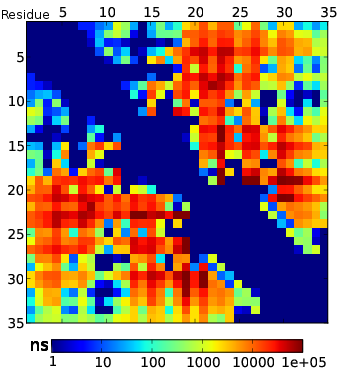
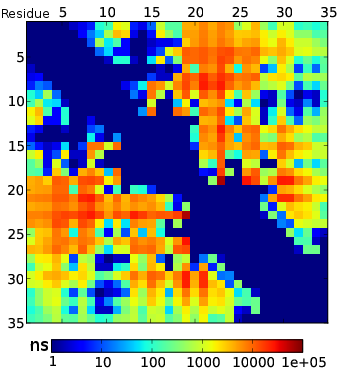
TTET Conclusions
- Experimental advances revealed multi-state kinetics in smallest protein system
- MSMs capture multi-state kinetics and recapitulate TTET measurements
- Detailed comparison of simulation and experiment reveals need for better forcefields and observables
Inferring Conformational Ensembles from Noisy Experiments (and simulation)
What if the force fields are broken?

Experiments as Projections

Experiments as Projections

Experiments as Projections

Two Models, Same "Data"


A Third Possibility
Ambiguous Measurements
Bayesian Energy Landscape Tilting
- Infer conformational ensembles from simulation and ambiguous experiments
- Simultaneously model structure and population
- Characterize posterior through MCMC
- Error bars on equilibrium and structural features
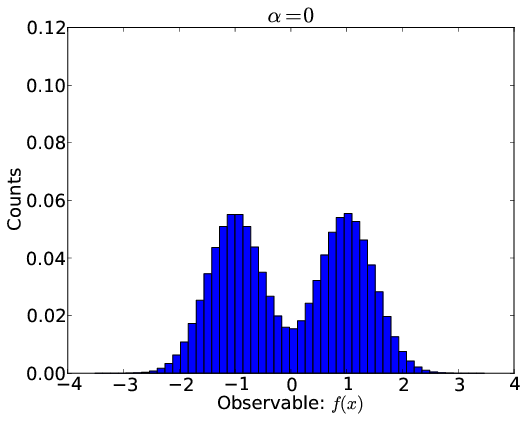

Ingredients for Ensemble Inference
- Equilibrium molecular dynamics simulation, with conformations $x_j$
- Equilibrium experimental measurements $F_i$ with uncertainties $\sigma_i$
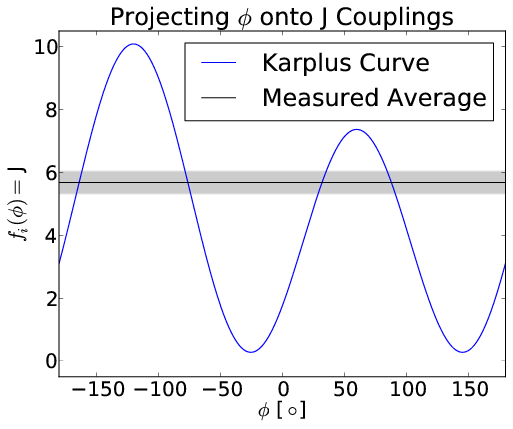
- Predicted experimental observables: $f_i(x)$
Biasing and Reweighting
- Project onto basis of predicted experimental observables
- Reweight using a linear free energy: $-\sum_i \alpha_i f_i(x_j)$
- $\alpha_i$ tells how populations are perturbed by $i$th experiment
Biasing and Reweighting
- Project onto basis of predicted experimental observables
- Reweight using a linear free energy: $-\sum_i \alpha_i f_i(x_j)$
- $\alpha_i$ tells how populations are perturbed by $i$th experiment
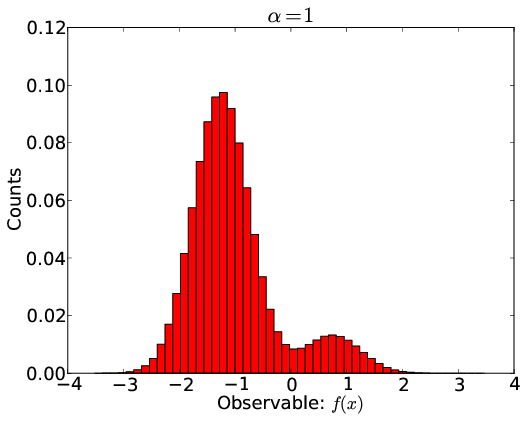
Biasing and Reweighting
- Project onto basis of predicted experimental observables
- Reweight using a linear free energy: $-\sum_i \alpha_i f_i(x_j)$
- $\alpha_i$ tells how populations are perturbed by $i$th experiment
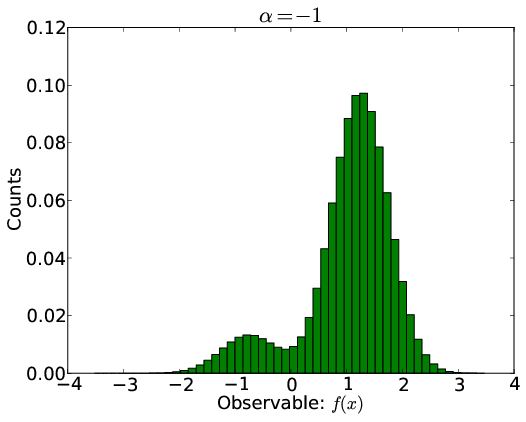
A Bayesian Framework
Assume independent normal errors.
$\underbrace{\log P(\alpha| F_1, ..., F_n)}_{Log _\ Posterior}$ = $\underbrace{-\sum_i \frac{1}{2}\frac{(\langle f_i(x)\rangle_α - F_i)^2}{\sigma_i^2}} _ {Log _\ Likelihood (\chi^2)} + \underbrace{\log P(\alpha)} _ {Log _\ Prior}$
Determine $\alpha$ by sampling the posterior.
FitEnsemble
An Open-Source Python Library for Ensemble Modeling
Trialanine
- Model for secondary structure / disorder peptides
- NMR Data: chemical shifts and scalar couplings
Idea: Use chemical shifts and scalar couplings to reweight simulations of trialanine performed in five different force fields.
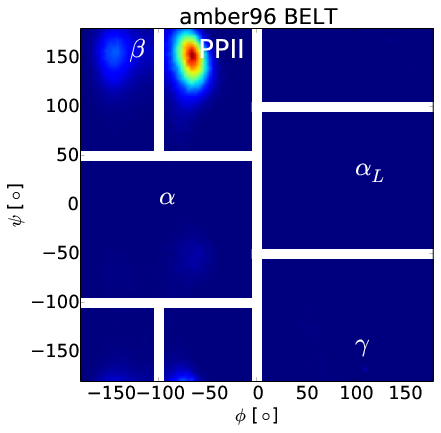
BELT Corrects Force Field Error
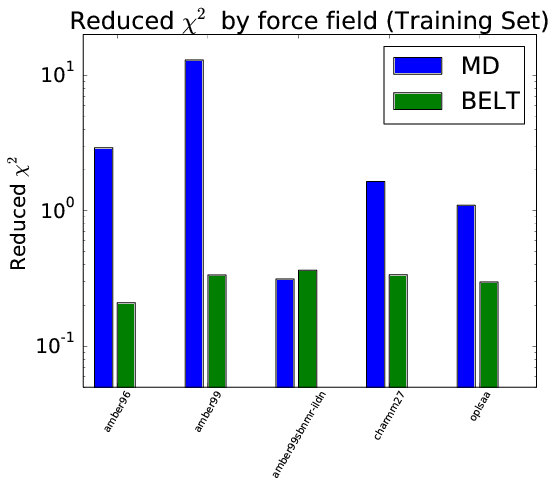
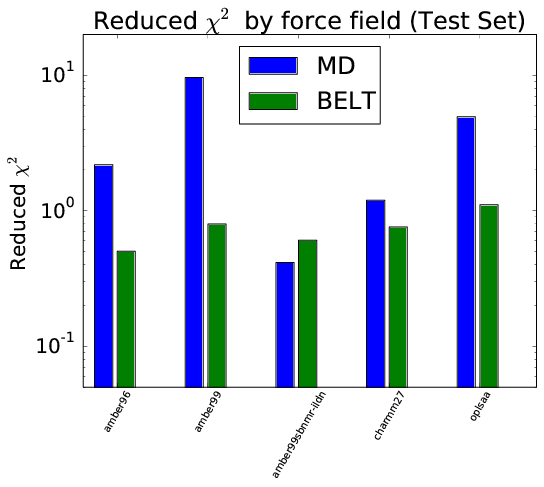
Robust to Over-Fitting
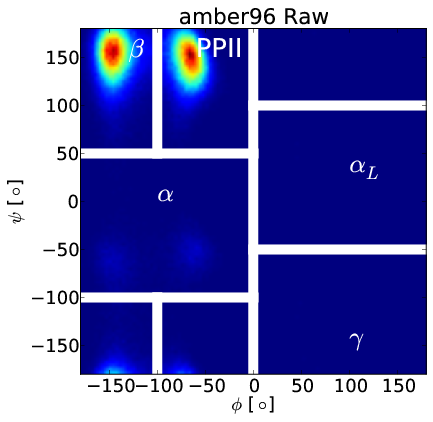
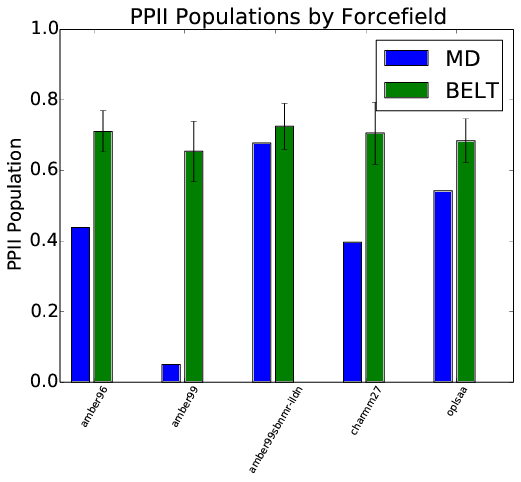
Correcting $\beta$ Bias in Amber96
Correcting $\beta$ Bias in Amber96
Forcefield-Independent Simulations
Bovine Pancreatic Trypsin Inhibitor
- Workhorse in protein folding, crystallography, and NMR
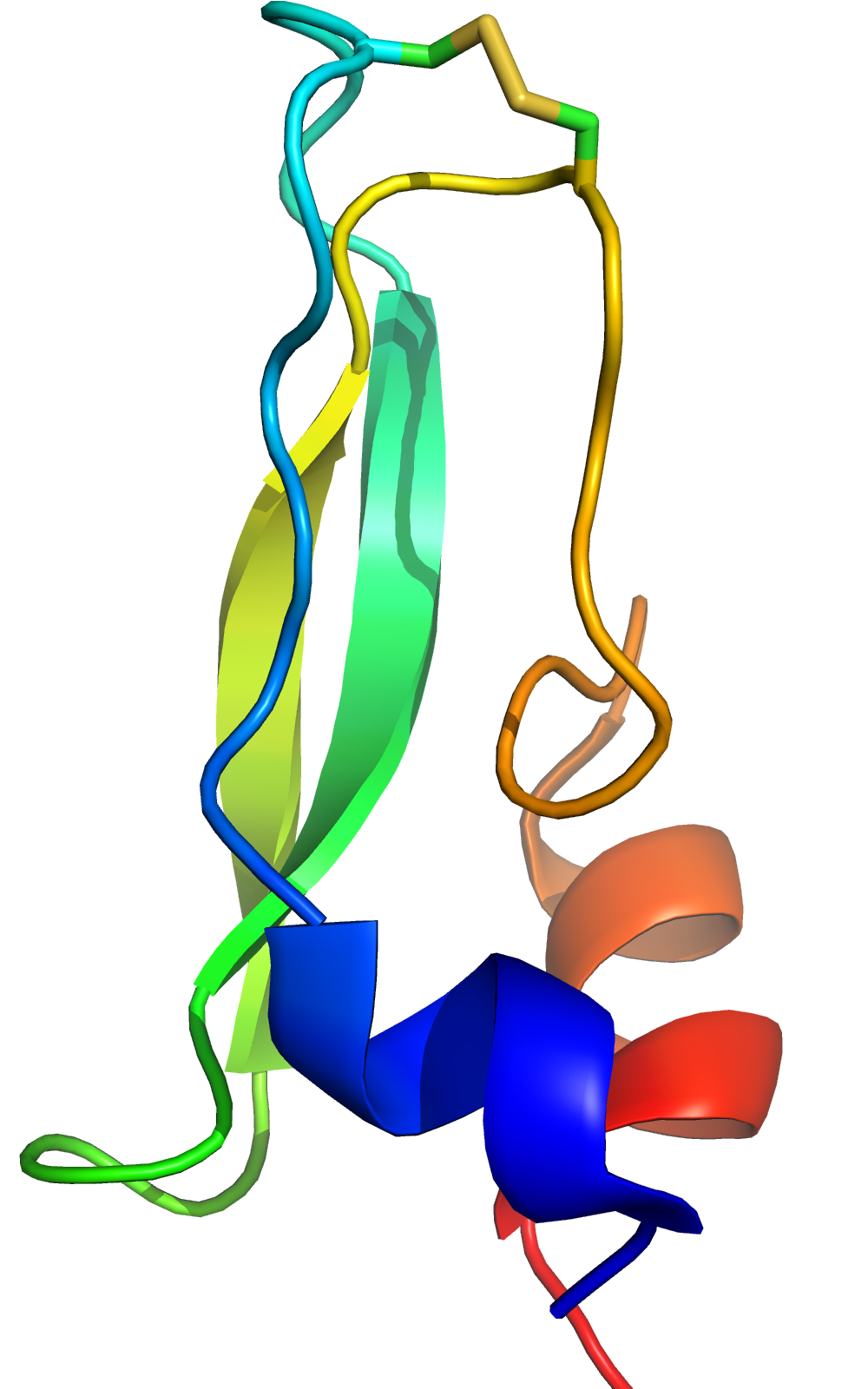
Bovine Pancreatic Trypsin Inhibitor
- Multiple disulfide (C14-C38) conformations in trypsin binding loop.
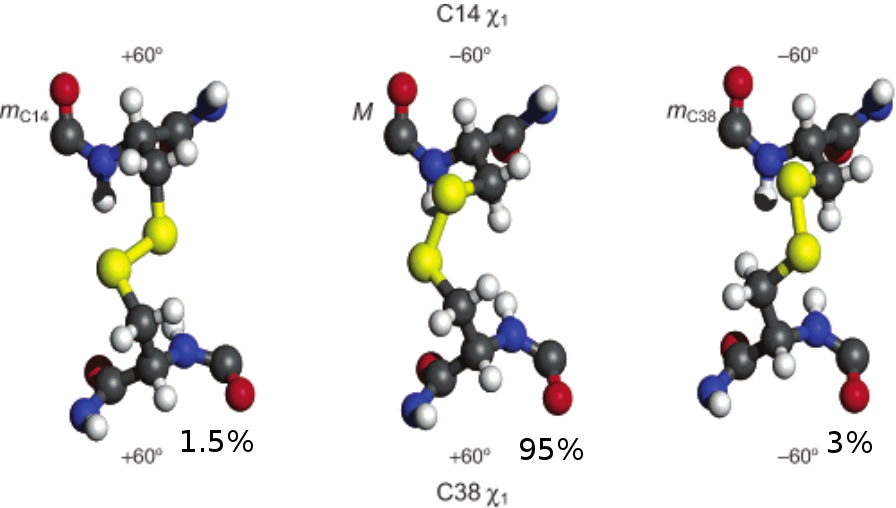
Are BPTI Simulations Consistent with Experiment?
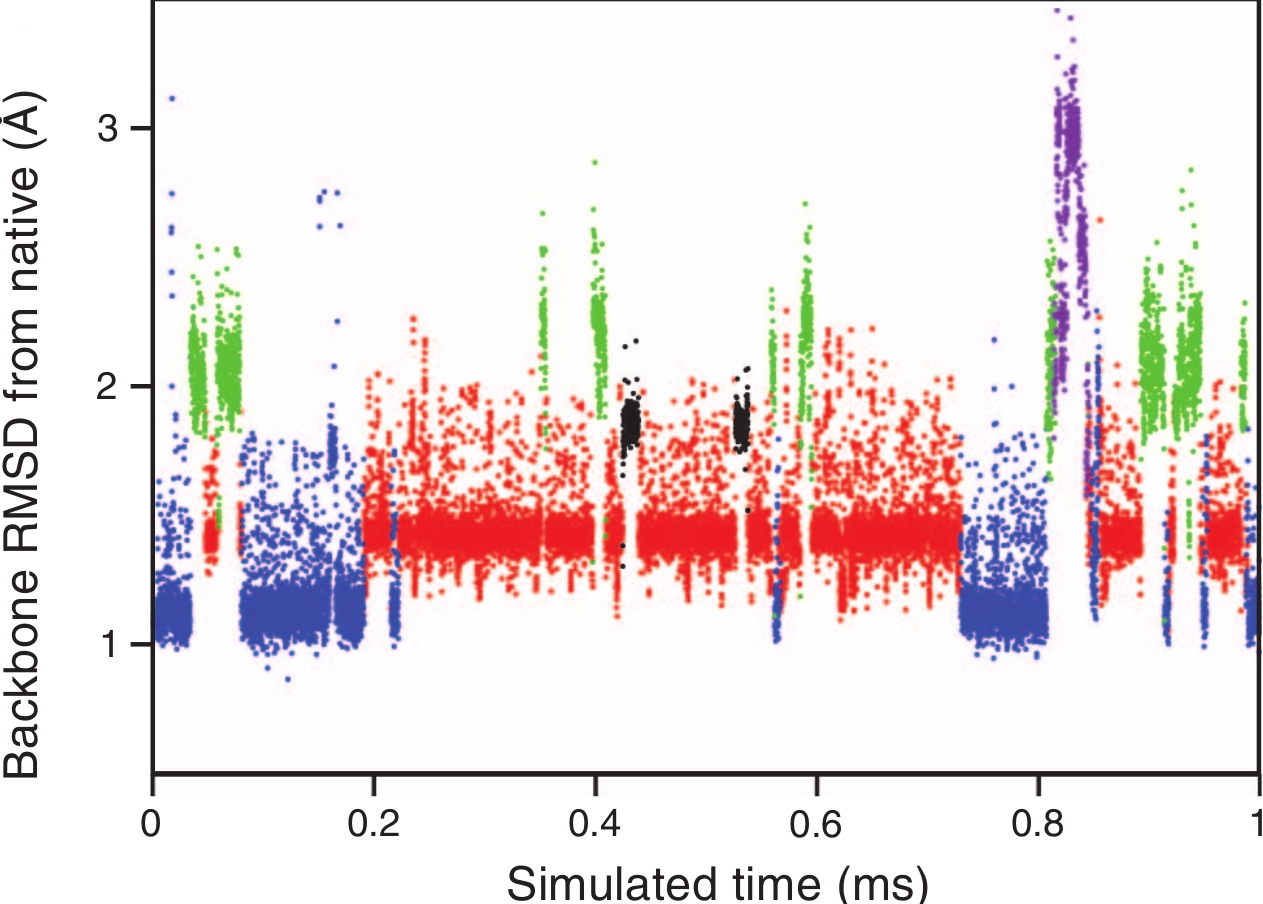
Simulation Favors Non-Native State
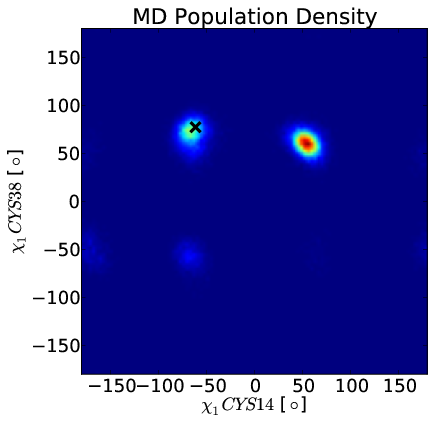
Idea: Use chemical shifts to reweight BPTI simulation.
BELT model Favors Native State
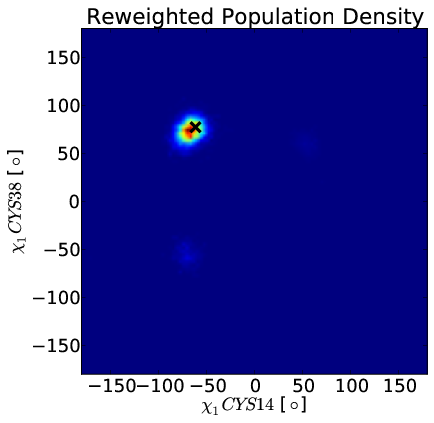
Models for BPTI
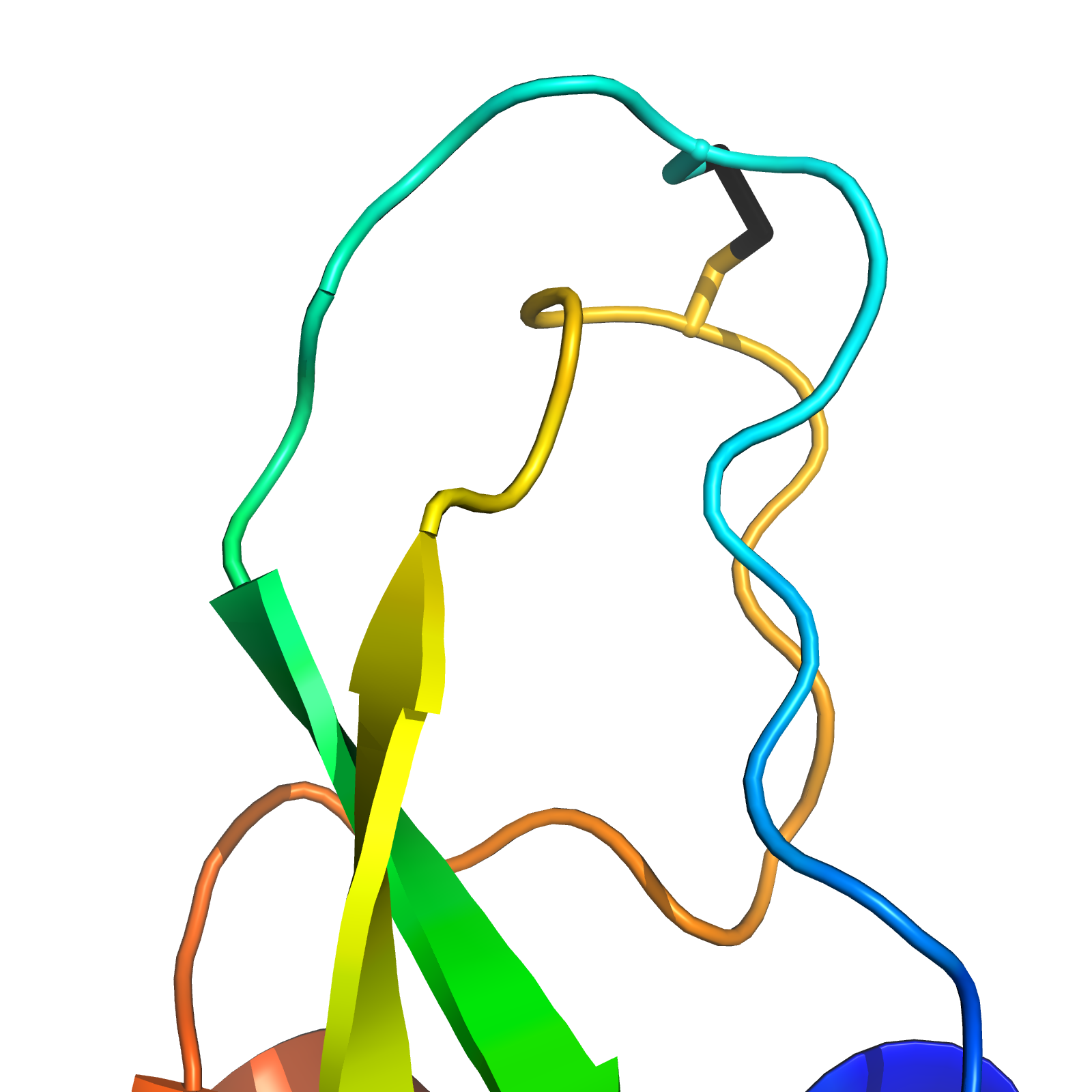
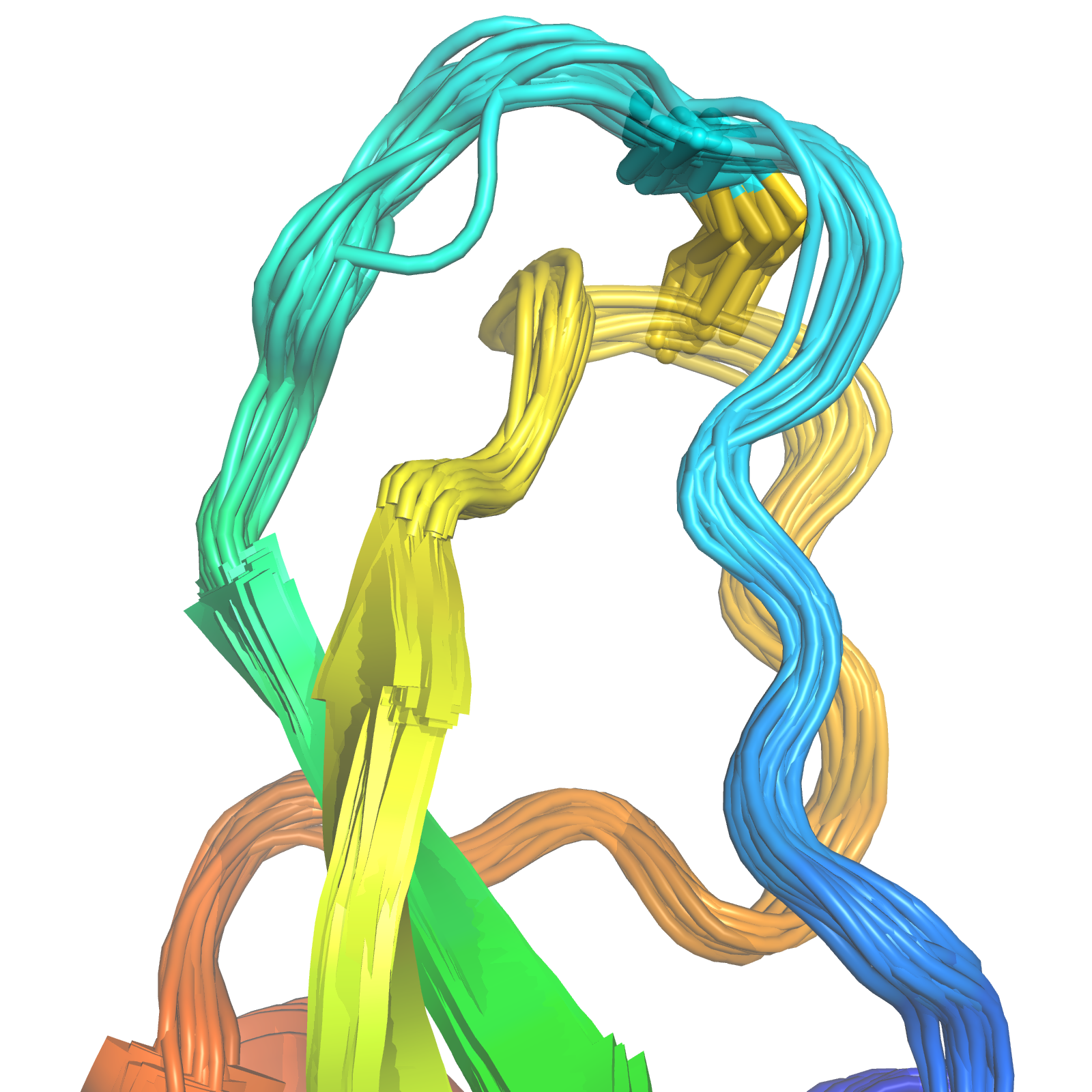
X-Ray MD BELT
Falsifying BPTI Models with J Couplings
$\frac{1}{n}\chi^2 = 15.0$ $\frac{1}{n}\chi^2 = 13.7$ $\frac{1}{n}\chi^2 = 10.4$
Conclusions and Future Work (BELT)
- BELT corrects forcefield bias in trialanine and BPTI
- Measure all scalar couplings for BPTI
- Better predictors of experimental observables: chemical shifts, scalar couplings, (TTET)
- Can we change the way NMR data is collected and analyzed?
Conclusion
- MSMs parallelize MD simulation, bringing millisecond-scale dynamics within reach
- Coupling MSMs to phenomenological model of TTET allows quantitative prediction of kinetic experiments
- BELT enables experiment-driven modeling of structure and equilibrium
- Open-Source Tools for protein inference (MSMBuilder, FitEnsemble)
Acknowledgements
Rhiju and Vijay
Acknowledgements
Pande Lab
Greg Bowman
Vince Voelz
Robert McGibbon
Christian Schwantes
TJ Lane
Imran Haque
Everyone!
Acknowledgements
Das Lab
- Pablo Cordero
- Frank Cochran
- Fang-Chieh Chou
- Parin Sripakdeevong
- Everyone!
Acknowledgements
Biochemistry / Biophysics
Buzz Baldwin
Dan Herschlag
Pehr Harbury
Xuesong Shi
Laura Wang, Jessica Metzger, Aimee Garza, Crystal Spitale and all the Biochem staff
Kathleen Guan
Everyone!
Acknowledgements
Committee
Todd Martinez
Pehr
Russ Altman
Acknowledgements
Folding@Home Donors and Forum Volunteers (Bruce Borden)
OpenMM Team: Joy Ku, Peter Eastman, Mark Friedrichs, Yutong Zhao
Thomas Kiefhaber, John Chodera, Frank Noe, Jesus Izaguirre
DE Shaw Research
Susan Marqusee, Laura Rosen
Thanks!
A Model for TTET
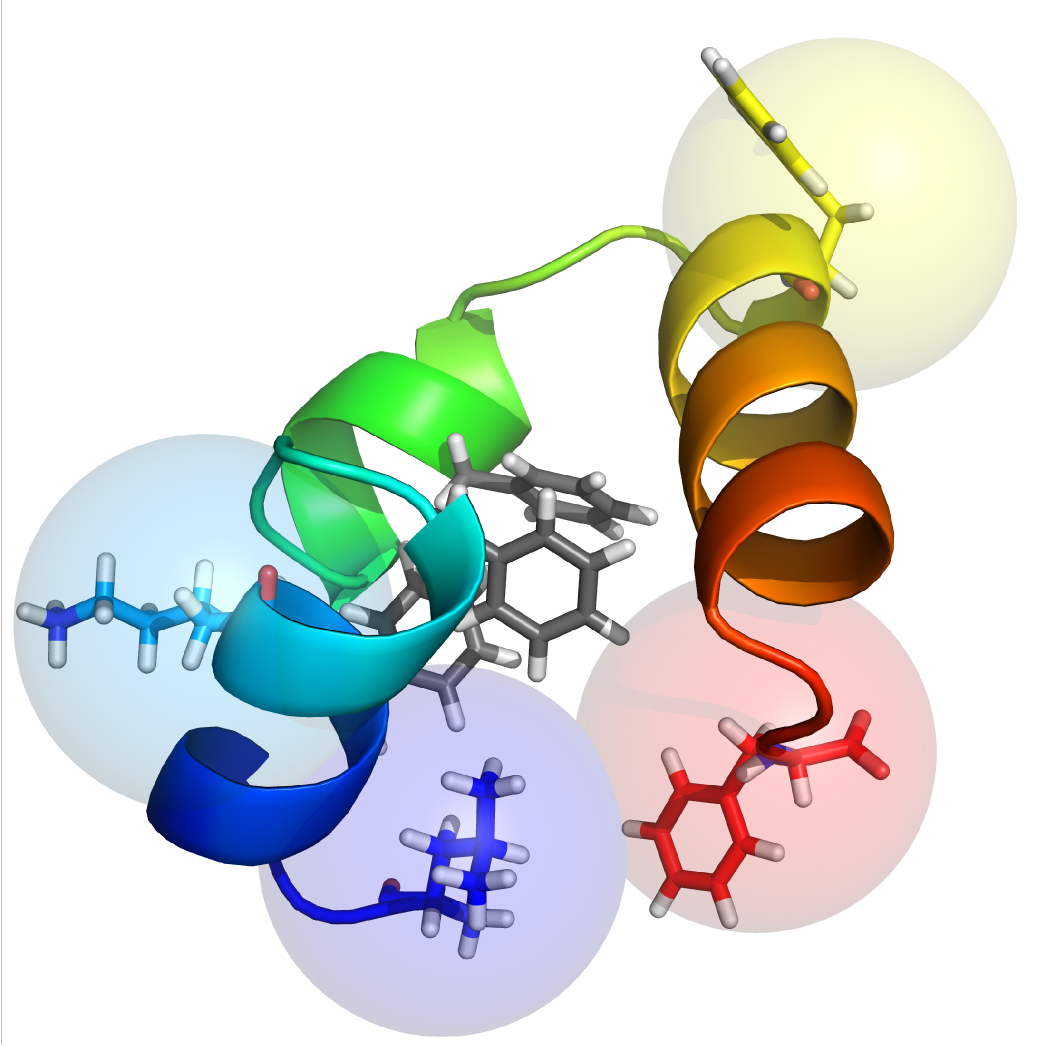
A Model for TTET
A Model for TTET
$$P(i\rightarrow j, dark \rightarrow dark) = P_0(i\rightarrow j)$$ $$P(i\rightarrow j, dark \rightarrow light) = P_0(i\rightarrow j)$$ $$P(i\rightarrow j, light \rightarrow light) = (1 - f_i) P_0(i\rightarrow j)$$ $$P(i\rightarrow j, light \rightarrow dark) = \delta_{ij} f_i$$
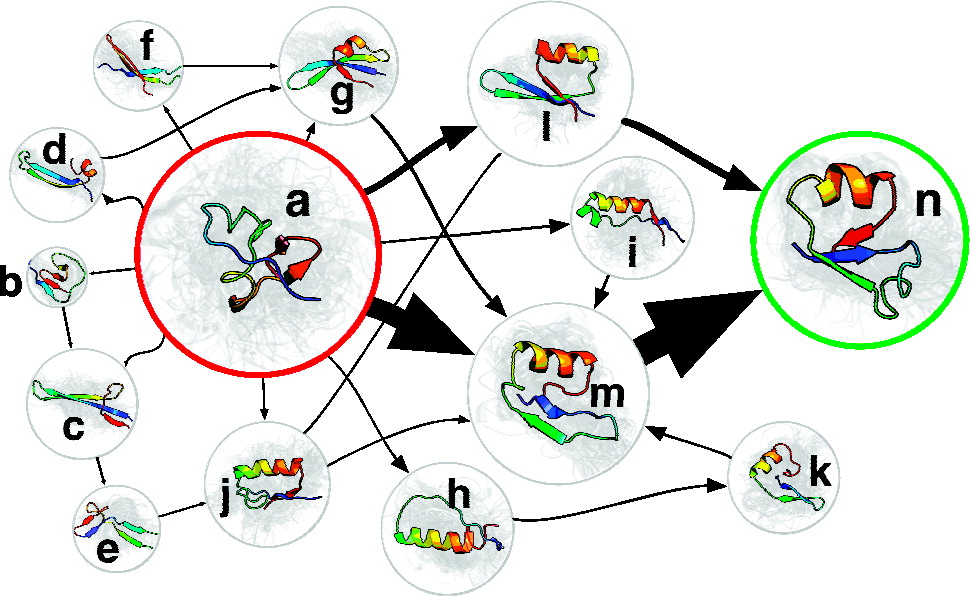
Conformational ensembles
- Structure + Equilibrium (not kinetics)
- Characterize the population of every conformation
- Rigorous connection to equilibrium measurements
Bayesian Energy Landscape Tilting (BELT)
Linear virtual biasing potential (LVBP): $\Delta U(x;\alpha) = \sum_i \alpha_i f_i(x)$
$\alpha_i$ tells how the energy of structure $x$ is changed by the predicted observable $f_i(x)$.
Populations by Boltzmann: $\pi_j(\alpha) \propto \exp[-\Delta U(x_j;\alpha)]$
A Bayesian Framework
The true ensemble is described by some coefficients $\alpha$.
Let $F_i$ be a noisy measurement of the $i$th experiment.
Let $\langle \rangle_\alpha$ denote an equilibrium expectation in the $\alpha$ ensemble.
$$P(F_i | \alpha) \approx N(\langle f_i(x)\rangle _\alpha, \sigma_i)$$
A Bayesian Framework
$$P(\alpha | F_1, ..., F_n) \propto P(F_1, ..., F_n | \alpha) P(\alpha)$$
$$\log P(\alpha| F_1, ..., F_n) = -\sum_j \frac{1}{2}\frac{(\langle f_j(x)\rangle _\alpha - F_j)^2}{\sigma_i^2} + \log P(\alpha)$$
MaxEnt prior: $\log P(\alpha) = \lambda \sum_i \pi_i(\alpha) \log \pi_i(\alpha)$
Use MCMC (pymc) to sample the likelihood function.